Recent advances in cell biology and genetic engineering have changed the way we think about medicine. Indeed, we are witnessing a potential shift from the traditional paradigm of health care to one with greater focus on the possibility of regenerative and curative treatments. Regenerative medicine (RM) can be defined as “self-healing through endogenous recruitment or exogenous delivery of appropriate cells, biomolecules and supporting structures” [1]. RM products are designed to address the underlying causes of diseases rather than just their symptoms.
Several kinds of RM products are just now beginning to reach late-phase trials and market authorization, and there are some companies that are advancing new chimeric antigen receptor (CAR) T cell-based therapies to Phase III clinical trials for several indications. Industry advocates hope that in the future, engineered T cells will be used to treat HIV, immune deficiencies, and autoimmune disorders, among numerous other conditions. To that end, some therapies have already been approved. For example, in 2015, the European Medicines Agency approved Holoclar®, an autologous stem cell-based therapy to restore the eyesight of patients with severe corneal damage. In 2016, Strimvelis, the first ex-vivo stem cell gene therapy using autologous CD34+ cells transduced to express ADA (adenosine deaminase), received European marketing authorization for treating ADA-SCID (severe combined immunodeficiency due to ADA deficiency). This year, Kite Pharma and Novartis will initiate a rolling submission of biologic license application (BLA) for CAR T cell therapy after demonstrating promising initial clinical data. On the tissue engineering front, the U.S. Food and Drug Administration (FDA) recently approved the first autologous cellularized scaffold product, a matrix-induced autologous chondrocyte implantation (MACI) to repair cartilage defects of the knee.
Today, more than 700 companies around the world make up the rapidly growing RM industry [2] and the global RM market is expected to grow to $67.6 billion by 2020, a large increase from $16.4 billion in 2013 [3]. These advances in RM are coming at a good time. The Centers for Medicare & Medicaid Services (CMS) project that health care costs will increase to $5.5 trillion and 19.9 percent of gross domestic product (GDP) by 2025 [4]. Some of this increase in health care costs will accrue because of palliative treatment of chronic diseases like heart disease, cancer, stroke, diabetes, and osteoporosis [5]. Advocates hope that RM treatments will help improve clinical outcomes and slow rising health care costs by offering effective curative therapies in place of palliative treatments for many of these chronic diseases.
In October 2016, the National Academies of Sciences, Engineering, and Medicine’s Forum on Regenerative Medicine (Forum) convened a 1-day workshop titled State of the Science in the Field of Regenerative Medicine: Challenges of and Opportunities for Cellular Therapies. The workshop brought together a broad range of researchers, clinicians, industry representatives, and patient advocates to discuss promises and challenges of translating RM to the clinic. Today, RM products include a broad array of emerging therapeutics including cell therapies (both stem cell therapies and immunotherapies), engineered tissues and organs, gene therapies, cell-based devices, and cell-free implants and materials that are designed to aid in tissue regeneration in vivo. The authors of this paper elaborate on the discussions that took place during the workshop and highlight areas of interest in preparation for the Forum’s next workshop on June 26, 2017, titled Navigating the Manufacturing Process and Assuring the Quality of Regenerative Medicine Therapies.
This paper is structured in a way that highlights some of the key aspects associated with cell therapy manufacturing, as discussed during the October 2016 workshop. It departs from an understanding that while there are currently an increasing number of products in the clinical pipeline and on the market, the industry has yet to coalesce around an appropriate cell sourcing, manufacturing, and logistics and delivery model, in part due to the rapid speed at which this sector evolves. Development of common platforms for crosscutting tools, measurement, and functional assays and a robust logistics and supply chain has undoubtedly advanced the translation of various products. The focus of the June 2017 workshop will be on manufacturing challenges associated with novel RM products. In this paper, the authors concentrate on three main aspects of RM manufacturing:
- Manufacturing: current manufacturing models, facilities, equipment, and analytical capabilities;
- Raw materials: cells, ancillary materials, and a discussion of quality attributes and parameters; and
- Regulatory environment (for definitions used throughout the discussion paper see Box 1).

Manufacturing: Current Manufacturing Models, Facilities, Equipment, and Analytical Capabilities
Models for Manufacturing Cell Therapies
The clinical administration of cell therapies, particularly allogeneic ones, has often relied on centralized and sometimes regional manufacturing models (e.g., some blood banks) [6]. In these models, a central site is responsible for the production of the cell therapy product (CTP), which is then delivered to the clinical site for use. Centralized manufacturing is attractive because it allows firms to achieve economies of scale and to ensure product consistency. Centralized manufacturing strategies have typically depended on a complex array of transport and logistical services including cryopreservation and specialized airfreight. As such, cryopreservation has become a critical area of interest in cell-therapy manufacturing, as issues of cell viability, functionality, and patient safety become increasingly important to resolve as more therapies reach the market. For autologous cell-based applications, many have argued that the best place for cell production is the “bedside” [7].
Automated tracking systems, chain-of-custody controls, and standardized quality assurance programs are critical to the generation of a robust and viable supply chain that bridges discovery, manufacturing, and administration. Alternatives to centralized manufacturing models have included strategies for cell production and processing at point-of-care, and various instruments enabling environmentally controlled, validated manufacturing of CTPs by the bedside. Hybrid models, which bridge multistep delivery with centralized manufacturing, have also appeared to gain some traction. Another viable option is regional “manufacturing hubs” that leverage key concepts of centralized manufacturing, yet are close enough to the bedside to allow live cell transportation. Significant technological advances are needed to (1) further improve cryopreservation and transportation methods to ensure minimal loss of therapeutic cell function during freezing, transit, and thawing, and (2) enable live-cell transportation without compromising quality and allowing continuous in-transit remote monitoring.
Both models can be pursued in tandem, and will each require optimization, the implementation of novel technologies, and industry-appropriate logistics to ensure that therapies are produced and delivered seamlessly and safely. Centralized manufacturing is a viable model for several products in the gene and cell therapy space as well as tissue-based products. For other therapies and RM applications, decentralized manufacturing may be more appropriate, although few models have been developed and implemented to date. Ultimately, issues of cell sourcing, raw material, and product quality control, automation, and the analytical footprint needed for robust and reproducible manufacturing will determine the appropriate model (see Figures 1 and 2 for a schematic of how these two manufacturing models are structured).

Figure 1 | Centralized Manufacturing Model. Source: Haddock, R., S. Lin-Gibson, N. Lumelsky, R. McFarland, K. Roy, K. Saha, J. Zhang, and C. Zylberberg. 2017. Manufacturing cell therapies: The paradigm shift in health care of this century. NAM Perspectives. Discussion Paper. National Academy of Medicine, Washington, DC. Note: Critical quality attribute (CQA)
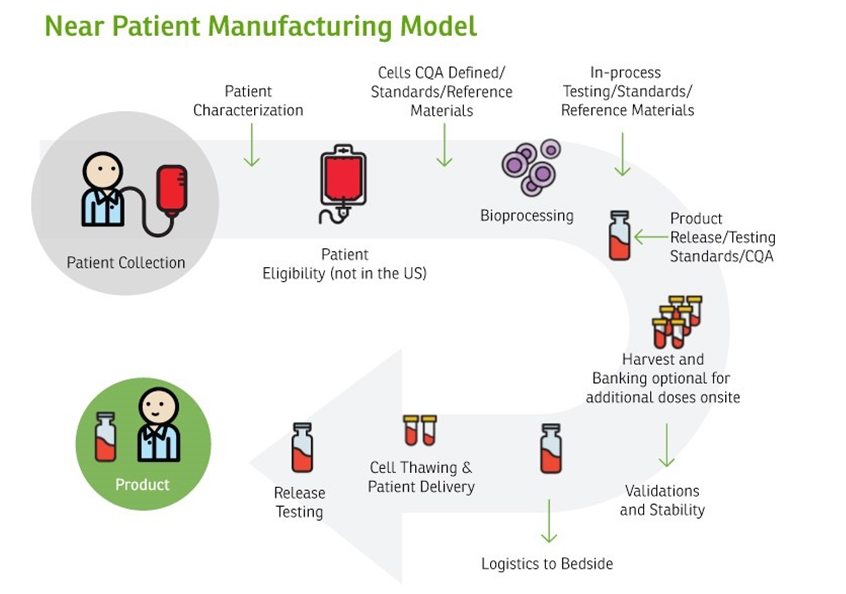
Figure 2 | Near Patient Manufacturing Model | Source: Haddock, R., S. Lin-Gibson, N. Lumelsky, R. McFarland, K. Roy, K. Saha, J. Zhang, and C. Zylberberg. 2017. Manufacturing cell therapies: The paradigm shift in health care of this century. NAM Perspectives. Discussion Paper. National Academy of Medicine, Washington, DC. Note: Critical quality attribute (CQA)
Firms developing RM therapies face several challenges in determining an appropriate manufacturing model for commercialization. Products that are not unique to the patient (e.g., allogeneic cell therapies, universal tissues) and have adequate stability to be shipped globally could be supplied via a single central manufacturing site, thus providing efficiency and consistency. However, products with limited stability, including certain cell types for autologous therapies, may require a distributed model in which manufacture occurs in multiple regional centers or are even co-located with treatment. A distributed model requires special consideration for consistency of manufacturing and analytical methods across sites and centralized oversight for data trending and release. Models reliant on shipment of RM products need to ensure validated, temperature-monitored, and timely transport that takes into account transport logistics and the potential for unforeseen delays. Firms planning international supply of RM products may benefit from working together and with government agencies to ensure these time-critical biological materials can be moved across international borders quickly, and new shipping and tracking technologies are shared and adopted. Manufacturing and regulatory standards need to be developed to ensure global harmonization.
Good Manufacturing Practice Facilities
Determining a specific strategy for the creation of a good manufacturing practice (GMP) facility for cell manufacturing would depend largely on the appropriate business model and the type of cell product. Whether bedside, a point-of-care manufacturing model (e.g., for autologous cells), a regional manufacturing hub model, or a centralized manufacturing model is most appropriate must be decided a priori based on patient needs, transportation and storage options, cost, and flexibility of the product. In addition to clinical manufacturing facilities, there is a great need for GMP-like facilities for product and process verification and validation as well as workflow simulation. It is difficult for industry or clinical centers alone to use their own GMP facilities for full-process validation or workflow establishment under GMP-like conditions; a possible solution to this issue is a model that relies on regional academic-industry-government consortiums and their facilities, such as those established by funding from the National Institute for Standards and Technology’s (NIST’s) National Cell Manufacturing Consortium (NCMC) or by the new Manufacturing Innovation Institutes cited in the next section.
Manufacturing Challenges and Opportunities
Recently NCMC, with funding from the NIST Advanced Manufacturing Technology Program (AMTech), convened industry, academia, and government experts to develop a 10-year technology roadmap for cell manufacturing. The master plan, Achieving Large-Scale, Cost-Effective, Reproducible Manufacturing of High-Quality Cells: A Technology Roadmap to 2025, outlines short-, medium-, and long-term technological advances that are needed for the successful advanced manufacturing of therapeutic cells and, more broadly, for growing the nascent industry [8].
In cell manufacturing, products are often “living” entities whose properties can change with even simple manipulations such as pipetting to transfer cells between culture dishes. Even in biologics manufacturing, where recombinant proteins or monoclonal antibodies are produced in animals or using cultured cells, the product (generally a protein) is still much simpler than a cell-based product and can be well characterized to ensure reproducibility.
For most cell-based therapies, more work must be done to identify what properties of a specific cell population make them effective or safe in a disease context. For example, current data indicate that the current set of surface markers used to identify cells as mesenchymal stromal/stem cells (MSCs), is insufficiently specific to ensure their functional quality [9,10]. More sophisticated attributes of cell quality are therefore critically needed [11].
The in-line and in-process measurement of critical quality attributes (CQAs) in real time and the development of quality by design (QbD) to ensure the highest quality and reproducibility of the manufacturing process are also necessary for the success of cell-based therapies. For translation into successful industry and clinical products, cell therapies must ensure that such CQA-driven quality manufacturing processes are in place, and that those processes are robust, reproducible, and cost-effective.
In addition to addressing the lack of regulatory standards in the field, there is an ongoing international effort to address standards of development for several aspects of the manufacturing of these novel products, including bioprocessing equipment, ancillary materials, cell counting, and transportation and logistics within International Organization for Standardization (ISO) initiatives, to name a few. Outcomes from these discussions may impact cost and quality attributes of released products and the delivery of those in specific geographic regions.
It is expected that application-specific and cell-specific CQAs will be identified through these international harmonization efforts. These will likely be multivariate discriminators identified through deep characterization of the therapeutic cell populations in the context of their in vitro behavior as well as in vivo function and performance (i.e., potency, efficacy, and safety profiles) in animal models and early clinical trials. Depending on the specific type of application, deep characterization would require, in addition to traditional surface markers, broad “-omics”-based measurements including genomics, epigenomics, transcriptomics, proteomics, secretome analyses, and metabolomics, when applicable.
In addition, biophysical characteristics and functions like adhesion properties, cell morphologies, cell migration behavior, etc., could also be part of multivariate CQAs. Once deep characterization is coupled with potency and safety, potentially through big data analytics and computational modeling tools, broad classifiers of “good” versus “bad” cells can be identified and used as CQAs. It should be noted that not all of these measurements/-omics would be necessary for any given cell product. It is possible that the CQAs could simply include quantitative descriptors; once a small set of CQAs or a set of quantitative classifiers have been identified, they can then be measured in-process or in real time during cell production (see “Sensors, In-Line Assays, and Automation” section below).
Equipment: Automation of Cell-Manufacturing Processes
Development of suitable manufacturing equipment and built-in controls for in-process testing remains a challenge. Processes are, in most cases, still open, with full manual processing in a classified environment, or semi-open, where some processing steps are manual and need a controlled room while others do not [8]. There is a push in the field to develop fully enclosed manufacturing systems that address not only the environment but also the in-process controls. Once manufacturing and controls are better understood, equipment specifications can be developed to better meet these requirements.
Investment in “flexible” automation will be important as the field continues to develop. As will be discussed in the “CQAs, critical process parameters (CPPs), and QbD” section, the variability of source-cell properties dictates that the manufacturing process be flexible and adaptable to ensure that the end products are of the same quality and consistency. This means that any automation of the biomanufacturing should ensure that culture conditions, length-of-culture, etc., can be controlled in-process, based on real-time measurements of CQAs, that is, be feedback-controlled. For allogeneic applications, cell banking and using donor cells that are already well characterized could minimize cell-source variability and require less flexibility, but feedback-driven process control would still provide improved consistency and product quality.
Sensors, In-Line Assays, and Automation
Innovations in automation will be important in the development of low-cost and reproducible manufacturing of CTPs. Variabilities in cell products can be introduced during manufacturing through inconsistent manual processing. Once CQAs and CPPs are established, bioprocessing tasks should be automated to ensure consistent CPPs and product quality across every batch and manufacturing centers, which will also help in reducing manufacturing costs in the long run.
Another important element for automation of the cell-manufacturing process will be the integration of in-line assays and measurements for CQAs as well as rapid measurements of potency, efficacy, and safety parameters. Despite the presence of many “sensors” in current bioreactor systems, most of them only measure culture conditions (e.g., pH, oxygen, glucose, and temperature) and a few metabolites that may not be directly indicative of CQAs. It is possible that CQAs for some applications and products would be multivariate and therefore in-line multiparametric measurements might be necessary, for example, -omics-based measurements or a combination of biochemical and biophysical property measurements. New innovations are critically needed in this field to develop such assays within closed-system manufacturing platforms and to make these assays work within the timelines and workflows of cell-therapy manufacturing.
Further, in addition to CQAs, rapid measurement of efficacy and safety would be of tremendous benefit. Although preclinical animal models are critical in establishing the potential for efficacy and safety of cell therapies during investigational product research and development, they are difficult to use in a product-manufacturing setting and they often have limited power to predict how human tissues will respond to a given cell therapy [12]. A potential way of ensuring quality in the context of human pathophysiology is the use of disease- or “organs-on-a-chip”-type platforms (or similar organoid models) that are being increasingly used by the pharma industry for drug discovery and toxicology applications [13]. For example, a sample of the product could be applied to in vitro organoid or on-chip disease/physiological models and tested to see whether the cells themselves or the metabolites/factors secreted by the product have the desired effects. These systems could also be used in understanding mechanisms of action and refining CQAs. Investment in this area may provide rapid surrogate assays, and it might be possible to integrate them in-line within the closed-system manufacturing platforms, which in turn may provide rapid or even real-time efficacy and safety measurements in human-organoid models. All of these chip/organoid models are in early stages of development, but outcomes need to be monitored carefully to recognize potential limitations in terms of measurements and data interpretation.
It should be recognized that post-administration to patients, specific cell subpopulations within the administrated cell pool can selectively proliferate/undergo apoptosis and/or expand/contract [14]. For example, profiling patients post-T cell infusion indicates that only a very small fraction of the T cell clones persisted in vivo [15]. These observations serve as a reminder that in vitro characterization of the cell population alone may not fully describe the phenotypes and genotypes of cells in the patient hours, days, weeks, and years after transplantation. In vitro characterization and CQAs should be refined continually based on feedback from patient data and improved understanding of mechanisms of action as patients are followed throughout and studied over many years. The concept of using the “patient as a bioreactor” to use and select/expand only the most effective therapeutic cells in vivo could lead to smaller numbers of cells necessary for infusions and thus significantly shorter manufacturing timelines and potentially low-priced products.
Analytics to Measure Critical Quality Attributes or Putative Critical Quality Attributes
Marked advances that have happened in big data analytics over the past few years may enable the identification of CQAs, which could then lead to the implementation of the QbD in cell manufacturing. Further, systems biology will play a role in identifying functionally relevant CQAs, and many efforts are already underway [16]. Comprehensive analyses of cell secretome, metabolome, surface markers, proteome, epigenome, and biophysical properties, along with analyses of cell efficacy, potency, and safety in animal models, organoid models, and human clinical trials, will enable systems biology approaches to establish robust CQAs. Ultimately, developing flexible bioprocesses specific for each cell type and each application that ensure maximizing of relevant CQA parameters may lead to QbD. This will also ensure minimal batch failures, predictability, consistent product quality, and high safety, efficacy, and potency, all of which would lead to increased success and lower cost of cell therapies.
Analytical methods that produce high confidence data underpin the decision-making process from research and development (R&D) to regulatory submissions, including developing and establishing CQAs. Recognizing the need for more robust analytical methods, NIST, with the input of the industry, has recently developed a generalized framework for designing and conducting cell measurements that consist of the following elements: (1) clearly defined measures, that is, what physical property is being measured that embodies quality attributes, (2) well-designed measurements that are qualified and fit-for-purpose, (3) measurements with in-process controls that provide measurement assurance, and (4) appropriate documentation, reporting, and communication [17]. This general framework is being applied to cell counting and cell viability measurements essential for all CTPs. Functional assays and other cell-based assays that correlate with clinical responses are more complex to ascertain. Other measurement challenges for CTPs include identifying appropriate biomarkers, validating rapid-release tests, and the need for in-process monitoring. The recently created Manufacturing Innovation Institutes, coupled with standardization efforts, aim to address these challenges.
Raw Materials: Cells and Ancillary Materials, with a Discussion of Quality Attributes and Parameters
Cells: The Backbone of Successful Translation
Powered by scientific and technical advances, the promise of cell-based RM for repairing and replacing damaged tissues and organs is rapidly expanding. The process of bringing cell-based therapies to the bedside consists of multiple steps, beginning with basic science proof-of-principle work and initial cell characterization, continuing into preclinical validation, scale up, manufacturing and clinical trials, and culminating with commercialization and licensing steps. To achieve its goals, the RM industry will be able to learn from several available R&D and manufacturing paradigms such as those previously developed in the gene therapy and hematopoietic cell transplantation sectors, where injected cells are purified using previously validated functional assays and/or are minimally manipulated. However, for RM to succeed, the industry will need to develop and perfect new paradigms that address the challenge of obtaining functional, scalable populations of stem and progenitor cells that must comply with stringent regulatory demands of efficacy, potency, and safety.
A critical aspect of CQA- and QbD-based manufacturing is the quality of source cells. In autologous applications, there can be a high inherent variability of source cell properties. Even in allogeneic applications, cell properties will vary from donor to donor. Approaches such as “master” or “working cell-banking” and “super-donor” concepts can minimize such variability for allogeneic sources. Establishing CQAs of source cells, whether autologous or allogeneic, is an important step in ensuring quality products. This suggests that the intermediate manufacturing process (or any manipulation between acquiring source cells and product release) must be “flexible” and “adaptable” to accommodate the variability of source cells and provide consistent products of the same CQAs.
Cell sources for RM applications represent a significant challenge for current manufacturing. The cell sources may vary from primary fetal cells to fully differentiated cells of various types, and may be obtained from in vitro-expanded cell populations or primary tissues. For some manufacturing processes, cells must be pre-selected based on their phenotypic characteristics. The developmental maturity of cells used for transplantation will vary depending on cell type and therapeutic needs. For some applications, immature cells will be transplanted to allow them to complete their maturation in vivo; for other applications, the in vitro maturation stage will be more extensive. The broad range and complexity of cell sourcing and processing will demand the development of appropriate validation assays [18,19].
Additionally, it will be important to develop effective approaches for targeted cell delivery, optimizing cells’ survival and function in vivo, their integration with host cells and tissues, and long-term monitoring of cells’ safety, toxicity, and other possible cell-mediated adverse events.
Characterization of Gene-Modified Cells
Characterization of gene-modified cells has relied on established guidelines for gene therapies in multiple regulatory regions, but new challenges are emerging in the cell-manufacturing field as described below.
Genome-integrating approaches, like transposon and viral delivery, can disrupt endogenous gene function, leading to insertional oncogenesis, while the non-integrating nuclease-based editing of endogenous genes typically generates a wide spectrum of DNA sequence variants at the edited locus.
The extent of gene modification has increased considerably, as multiple rounds of modification are planned for emerging T cell therapies, including lentiviral gene delivery and TALEN or CRISPR-Cas gene editing. Viral vectors can integrate regardless of the multiplicity of infection into 10,000 different locations in the human genome, and expression of the viral transgene could be affected by the epigenetic status of each integration site. In addition, gene editing can generate hundreds of DNA sequence variants at the intended target site and unintended off-target site. Given that 10 million to 10 billion cells are typically modified during manufacturing, functional characterization of these modified cell populations is especially challenging in these multiplexed modification workflows [20, 21].
Characterization by next-generation sequencing typically provides a percentage of modified or edited alleles. Such characterization to date indicates a significant number of point mutations, translocations, and copy number variations throughout the genome for manufactured cells, even for targeted editing strategies [22]. Some of this variation can be due to the manufacturing process, or so called “culture-associated” genomic variation [23], and additional genetic variation can be due to off-target editing by nucleases. While DNA-targeting specificities of nuclease-based genome-editing agents are known, no simple algorithm to date can accurately and comprehensively predict off-target gene-editing effects [24]. Further, functional relevance of each modified allele is difficult to determine from sequencing data alone. To ensure quality, complementary cell functional assays should be performed for each application.
Raw Materials: The Complexity of the Starting Materials
Another source of variability in the manufacturing system is ancillary materials (AMs), or processing agents used to help ensure the safety of a therapeutic substance (in this case, cells). The potential source(s), magnitude and directionality of the factors generating this variability may not be evident early in a manufacturing development effort. In fact, it may only be discovered over time and may be multifactorial. This situation suggests that efforts to control the variability across the supply chains for AMs may be beneficial to controlling RM product variability. The source of AMs and adjoining requirements for suppliers and users have been the focus of standards development organizations (SDOs) like ISO and ASTM International, which have become more active in the RM field. Since most of the components used to manufacture AMs have different origins, both geographical and species-specific (due to worldwide restrictions on animal- and human-sourced materials), a common understanding of safety issues may be needed.
The complexity in manufacturing some of these materials brings challenges and illuminates the potential for updates and advances in standardization. The sourcing of AMs, as well as the common understanding of safety, is still a subject of continuing discussion. AMs require thorough understanding, which includes harmonization in their nomenclature and clarity with respect to regulatory guidance and regulations. Control and harmonization efforts for release and compliance have been described in several publications and SDOs’ efforts, such as the U.S. Technical Advisory Group to International Organization for Standardization Technical Committee 276 WG4 Bioprocessing on AMs for users and suppliers. This and similar efforts are still ongoing nationally and internationally.
Critical Quality Attributes (CQAs), Critical Process Parameters (CPPs), and Quality by Design (QbD)
Manufacturing of biopharmaceutical or cell-based medicinal products is generally defined as CQA- and CPP-designed to ensure product safety and efficacy. Ideal CTPs should demonstrate the desired clinical outcome (i.e., effectiveness) with little or no adverse reactions to patients (i.e., safety). In the United States, FDA defines a CQA as “a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality” [25]. Each CTP will likely have its own set of CQAs based on the product’s intended use and/or mechanism of action. The safety and efficacy of a CTP also hinges on the ability to consistently manufacture the product, that is, from batch to batch and across manufacturing centers, regardless of the scale of production. It is therefore important to establish CPPs whose variability impacts CQAs. CPPs should be monitored or controlled to ensure that the manufacturing process leads to products of desired quality.
Several approaches, including QbD, are actively being evaluated as tools for improving the quality of products and processes. QbD is a scientific and risk-based approach that involves deliberate design effort from product conception through commercialization [25]. An important requirement for QbD implementation is a full understanding of how product attributes and processes are related to product performance. Although CQAs are not well understood for many potential cell therapy products, some principles of QbD may be useful to improve manufacturing quality.
As shown in Figures 1 and 2, cell manufacturing involves a number bioprocessing steps that are sometimes referred to as “needle-to-needle processing,” that is, from collecting cells from a patient/donor to administering the cell product to patients. Each upstream process, such as cell collection, separation, purification, genetic modification, and expansion or differentiation, as well as each downstream process, such as harvesting, washing, purification, batch release, packaging, and shipping, can be further divided into specific steps, or “unit operations.” A robust manufacturing requires that each unit operation throughout the manufacturing process has in-process controls and measurements to achieve quality and consistency [26]. CQAs may be considered quality attributes tied to the intended clinical response. Characterization of the starting cell materials to the CTP is an important consideration for advancing R&D to manufacturing. CTPs can be characterized via a range of in vitro assays, include immunocytochemistry or immunohistochemistry, flow cytometry, and gene expression analyses, as well as other viability and functional assays. Appropriate functional assays that help to assess the likelihood of clinical success should be included as early as is feasible in the product development process. The selected assays will ideally be robust, technically simple, and high-throughput to enable rapid screening of potential targets. For allogeneic cell-based therapies, it may be necessary to consider the compatibility of immunological parameters of the transplanted cells and the host.
Manufacturing CTPs of defined CQAs requires control of the starting cell populations, AMs, equipment, and other bioprocessing. Robust analytical methods, including in-process monitoring and release assays, may help in advancing preclinical studies and clinical trials. Appropriate standards, quality control protocols, and standard operating procedures for all cell manipulations should be in place before large-scale translation of RM therapies commences.
CQAs are established by a given CTP intended for defined clinical indications. It is important to note that our understanding of the science is incomplete and little is known about the mechanism of action and quality attributes indicative or predictive of clinical efficacy for most indications. The role of fundamental basic research to elucidate these aspects should not be overlooked. In the meantime, studies can be designed to contribute to the broad understanding of CQAs and clinical efficacy.
Regulatory Challenges
As described in previous sections, RM products are typically orders of magnitude more complex than traditional small molecule drugs, recombinant protein drugs, and traditional implantable medical devices. In addition to their inherent compositional complexity, these products have significant heterogeneity and uncertainty in terms of mechanism(s) of clinical activity and safety profile. These sources of uncertainty taken together with the relative immaturity of the field naturally generate a desire for more guidance from regulators; however, premature prescriptive and/or proscriptive regulatory approaches have the potential to forestall further innovation because neither regulators nor developers can anticipate how science- and discovery-driven innovation will shape future products. Regulatory authorities in major pharmaceutical regions have endeavored to use all of the available regulatory tools, often through innovative interpretations, to fulfill their country-specific legislative mandates to protect the health of their respective publics. The topics discussed in the current paper highlight the existing tension between regulatory certainty and innovation.
Current Good Manufacturing Practice (cGMP) provides a general regulatory framework to ensure quality of RM products. The principles embodied in cGMP include the following components: exerting process control, testing product quality attributes to meet specifications, and ensuring consistency and reproducibility of each production run. Application of these cGMP principles has been well established in manufacturing of small molecules and biologic drugs, but application of these cGMP principles to RM products is frequently more challenging than a simple “cut and paste” exercise due to the complexities of RM products and their manufacture, as exemplified in this section.
Autologous Versus Allogeneic and Scale Out Versus Scale Up
For autologous products, patients’ own cells are collected and manipulated before being injected back for therapeutic effect (see Figure 1). As each patient’s cellular composition and physiological states differ, cells sourced from an individual can vary significantly for an idealized cell donor. Therefore, maintaining control becomes a significant challenge from the start of the manufacturing process. When each patient’s cell production is considered one batch, setting arbitrarily tight specifications for product release to meet an idealized cGMP requirement is difficult.
In addition, the traditional model of scale-up of a production process, which includes generating bigger batches of therapeutic products from a single source of starting material, does not function well for an autologously sourced production. In this case, expanding to an industrial-scale production process must follow a scale-out model in which, in order to expand in scale rather than producing more cells in each batch, more batches must be made, one (or a few) for each patient. The variability inherent in the starting materials from each patient will always exist so the scale-out process may need different control strategies than the scale-up process to achieve manufacturing consistency. In this scenario, methods for defining and measuring consistency may require additional exploration.
Target Audience of cGMP Regulations
For biopharmaceutical products, compliance with cGMP regulations occurs in a well-defined manufacturing facility that is subject to inspections for product licensure. For RM products, the traditional boundary of a single or a few manufacturing facilities under cGMP oversight may not be applicable, and exploration of new approaches to accommodate different manufacturing and oversight structures that provide similar product consistency and patient safety attributes of traditionally manufactured products may be warranted. For example, when cells are collected from donors, or patients themselves, and cellular product manufacturing is conducted near patient facilities such as clinics rather than at traditional manufacturing facilities, the personnel who perform such procedures are typically clinical professionals who may not be trained to comply with cGMP regulations. The challenge also exists at the tail-end of the cell therapy workflow, after products are released from the manufacturing facility and shipped to clinical sites for administration. As maintaining product quality is critical throughout the entire workflow, ensuring application of cGMPs, or comparable standards, at both ends of the workflow has become a significant challenge.
Process Change and Comparability Assessment
Manufacturing changes are commonplace during drug development and post-marketing for various purposes, such as scaling and process optimization. RM manufacturing is no exception. In addition, since the RM field is relatively new, many technologies available to manufacture RM products are not as advanced. As innovative technologies become available to improve the manufacturing process, changes are inevitable and meant for process improvement. The current regulatory framework requires that manufacturers conduct comparability assessments, which generally encompass analytical evaluations, at minimum, to demonstrate that the new process can generate products that meet the same established CQA specifications as the old process. Due to the biological nature of RM products at large, defining relevant CQAs and setting reasonable acceptance criteria are challenging. Using these CQA specifications becomes even more difficult when it is necessary to govern a presumably improved process incorporating new manufacturing technologies. Determining how to evaluate manufacturing changes with appropriate comparability assessment warrants further discussion.
In Need of Standards and Regulatory Science
Translating cGMP principles to actionable metrics is often achieved through use of standards, both regulatory and consensus. The need for standards has been voiced from major stakeholders in the RM space [27]. Standards are “common and repeated use of rules, conditions, guidelines, or characteristics for products or related processes and production methods, and related management systems practices” [28]. In the arena of regulated medicinal products, reference standards and highly characterized physical specimens are also critical and closely tied with the documented U.S. Pharmacopeial (USP) Reference Standards. Standards that are developed to fill specific niches of need for standard approaches and/or references addressing frequently used analytical steps, or manufacturing steps in RM manufacturing, can complement regulations, frame the pathway for innovation, improve efficiency, decrease costs, and increase consumer confidence in each final product, and in turn, the field as a whole.
Standards must be developed with a clear scope, purpose, stakeholders, and possible intended and unintended consequences in mind. Developing standards also requires a field to have achieved a certain maturity, a state that RM is rapidly approaching. With this maturity comes a sufficient level of community knowledge to feed the well-established and recognized, but rigorous, process used by American National Standards Institute (ANSI)-accredited SDOs to develop standards. Additional information on standards development may be found at the International Organization for Standardization.
As the RM field is advancing rapidly, the need for standards becomes more evident. Those standards will be needed to streamline manufacturing processes and optimization. Innovative thinking for both industry and regulators is also important. Some suggestions for the industry to consider include developing consensus papers or best practices that will eventually progress into standards. For regulators, promoting regulatory science in developing new tools and approaches, particularly regarding quality assessment of RM products, could help advance the field. By doing so, all stakeholders can work together to build a knowledge database to advance standards development and regulatory science. The newly formed Standards Coordinating Body (SCB) for Gene, Cell, and Regenerative Medicines and Cell-Based Drug Discovery is organized as a consortium of nongovernmental stakeholders that operates through public–private partnerships with government regulatory agencies and other government organizations involved in helping to establish consensus standards for RM products. As evidence of recognition of this need, the recently passed 21st Century Cures Act recognizes the need for standards development in RM in Section 3036.
Looking Forward
It is expected that the upcoming manufacturing workshop hosted by the Forum on Regenerative Medicine on June 26, 2017, will open the discussion around decentralization or centralization modalities. In the case of allogeneic products, we can anticipate that after the cell product (drug) order is received, the central manufacturing site can ship the banked cells to the regional/local sites for thawing/expanding/filling/finishing and testing for release. The discussions that will be at the heart of the next workshop will revolve around questions of cell characterization as starting material, cell validation as a finished product, and manufacturing challenges both in bioprocessing and analytical testing. These essential parts—bioprocessing and analytical testing—are needed to obtain an industrial process that is reproducible and traceable.
The workshop will also address current and future manufacturing models best suited to bring these products to market in a scalable, safe and cost-effective manner that fits within a regulatory framework with standardized parameters. For example, for autologous manufacturing, the apheresis step/cell collection from patients can be conducted at regional/local sites, or, perhaps the produced cells can be frozen to be shipped to the local site. In the case of CAR-T cell manufacturing, processes can be performed in stages and at different locations. The workshop will address the role of cell banking as part of the process flow, either at initial stages (master cell banks), in-process byproducts, or as a released product. It is a key activity in the process flow and a known area from the standards and regulatory perspective. Advances and gaps in terms of analytical methods, measurements, and standards will also be part of the agenda. Several examples will be discussed as potential models applicable for this industry.
The upcoming workshop will open the discussion and the opportunity to look for new areas of research and advancement in the implementation of these new medicinal approaches. Among other objectives, this workshop will bring an understanding of what could be done with an industrialized mindset and a regulated approach to continue to fulfill the unmet medical needs of many patients today and tomorrow.
Join the conversation!
![]() Tweet this! Advances in genetics & cell biology are changing how we think about medicine. A paradigm shift in health care: http://bit.ly/2tRmAOv
Tweet this! Advances in genetics & cell biology are changing how we think about medicine. A paradigm shift in health care: http://bit.ly/2tRmAOv
![]() Tweet this! Regenerative medicine is a whole new realm of healing. Challenges & opportunities for this growing market: http://bit.ly/2tRmAOv
Tweet this! Regenerative medicine is a whole new realm of healing. Challenges & opportunities for this growing market: http://bit.ly/2tRmAOv
![]() Tweet this! Regenerative medicine is a game changer, but bringing these therapies to the bedside is complicated. Here’s a look: http://bit.ly/2tRmAOv
Tweet this! Regenerative medicine is a game changer, but bringing these therapies to the bedside is complicated. Here’s a look: http://bit.ly/2tRmAOv
Download the graphic below and share it on social media!

References
- Multi-Agency Tissue Engineering Sciences (MATES) Interagency Working Group. 2007. Advancing tissue science and engineering: A foundation for the future. Available at: https://webarchive.library.unt.edu/eot2008/20080916012251/http://tissueengineering.gov/advancing_tissue_science_&_engineering.pdf (accessed June 21, 2017).
- ARM (Alliance for Regenerative Medicine). 2016. Industry snapshot. Available at: https://alliancerm.org/page/industry-snapshot (accessed May 8, 2017).
- Sumant, O. 2014. Regenerative medicines market by technology (cell therapy, gene therapy, tissue engineering, small molecules and biologics), material (biodegradable synthetic polymers, scaffold, hydrogel and collagen, transgenic, fibroblasts, small molecules and biologics)—Global opportunity analysis and industry forecast, 2014–2022. Allied Market Research. Available at: https://www.premiummarketinsights.com/reports-amr/regenerative-medicines-market (accessed August 25, 2020).
- CMS (Centers for Medicare & Medicaid Services). 2017. National health expenditure data—Projected. Available at: https://www.cms.gov/research-statistics-data-and-systems/statistics-trends-and-reports/nationalhealthexpenddata/nationalhealthaccountsprojected.html (accessed May 8, 2017).
- Bipartisan Policy Center. 2012. What is driving U.S. health care spending? Available at: http://www.rwjf.org/content/dam/farm/reports/issue_briefs/2012/rwjf401339 (accessed May 8, 2017).
- Terzic, A., M. A. Pfenning, G. J. Gores, and C. M. Harper, Jr. 2015. Regenerative medicine build-out. Stem Cells Translational Medicine 4(12):1373–1379. https://doi.org/10.5966/sctm.2015-0275
- Kaiser, A. D., M. Assenmacher, B. Schroder, M. Meyer, R. Orentas, U. Bethke, and B. Dropulic. 2015. Towards a commercial process for the manufacture of genetically modified T cells for therapy. Cancer Gene Therapy 22(2):72–78. https://doi.org/10.1038/cgt.2014.78
- National Cell Manufacturing Consortium. 2016. Achieving large-scale, cost-effective, reproducible manufacturing of high-quality cells: A technology roadmap to 2025. Available at: http://www.cellmanufacturingusa.org/sites/default/files/NCMC_Roadmap_021816_high_res-2.pdf (accessed May 17, 2017).
- Lo Surdo, J. L., B. A. Millis, and S. R. Bauer. 2013. Automated microscopy as a quantitative method to measure differences in adipogenic differentiation in preparations of human mesenchymal stromal cells. Cytotherapy 15(12):1527–1540. https://doi.org/10.1016/j.jcyt.2013.04.010
- Mendicino, M., A. M. Bailey, K. Wonnacott, R. K. Puri, and S. R. Bauer. 2014. MSC-based product characterization for clinical trials: An FDA perspective. Cell Stem Cell 14(2):141–145. https://doi.org/10.1016/j.stem.2014.01.013
- Sacchetti, B., A. Funari, S. Michienzi, S. Di Cesare, S. Piersanti, I. Saggio, E. Tagliafico, S. Ferrari, P. G. Robey, M. Riminucci, and P. Bianco. 2007. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 131(2):324–336. https://doi.org/10.1016/j.cell.2007.08.025
- FDA (U.S. Food and Drug Administration). 2013. Guidance for industry: Preclinical assessment of investigational cellular and gene therapy products. Available at: https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM376521.pdf (accessed May 9, 2017).
- Bhatia, S. N., and D. E. Ingber. 2014. Microfluidic organs-on-chips. Nature Biotechnology 32(8):760–772. https://doi.org/10.1038/nbt.2989
- Di Stasi, A., S. K. Tey, G. Dotti, Y. Fujita, A. Kennedy-Nasser, C. Martinez, K. Straathof, E. Liu, A. G. Durett, B. Grilley, H. Liu, C. R. Cruz, B. Savoldo, A. P. Gee, J. Schindler, R. A. Krance, H. E. Heslop, D. M. Spencer, C. M. Rooney, and M. K. Brenner. 2011. Inducible apoptosis as a safety switch for adoptive cell therapy. New England Journal of Medicine 365(18):1673–1683. https://doi.org/10.1056/NEJMoa1106152
- Chapuis, A. G., C. Desmarais, R. Emerson, T. M. Schmitt, K. C. Shibuya, I. P. Lai, F. Wagener, J. Chou, I. M. Roberts, D. G. Coffey, E. H. Warren, H. Robins, P. D. Greenberg, and C. Yee. 2017. Tracking the fate and origin of clinically relevant adoptively transferred CD8+ T cells in vivo. Science Immunology 2(8). https://doi.org/10.1126/sciimmunol.aal2568
- Peltier, J., and D. V. Schaffer. 2010. Systems biology approaches to understanding stem cell fate choice. IET Systems Biology 4(1):1–11. https://doi.org/10.1049/iet-syb.2009.0011
- Lin-Gibson, S., S. Sarkar, and Y. Ito. 2016. Defining quality attributes to enable measurement assurance for cell therapy products. Cytotherapy 18(10):1241–1244. https://doi.org/10.1016/j.jcyt.2016.07.002
- Chen, K. G., B. S. Mallon, R. D. McKay, and P. G. Robey. 2014. Human pluripotent stem cell culture: Considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 14(1):13–26. https://doi.org/10.1016/j.stem.2013.12.005
- Fox, I. J., G. Q. Daley, S. A. Goldman, J. Huard, T. J. Kamp, and M. Trucco. 2014. Stem cell therapy. Use of differentiated pluripotent stem cells as replacement therapy for treating disease. Science 345(6199):1247391. https://doi.org/10.1126/science.1247391
- Wu, J., A. Platero-Luengo, M. Sakurai, A. Sugawara, M. A. Gil, T. Yamauchi, K. Suzuki, Y. S. Bogliotti, C. Cuello, M. Morales Valencia, D. Okumura, J. Luo, M. Vilarino, I. Parrilla, D. A. Soto, C. A. Martinez, T. Hishida, S. Sanchez-Bautista, M. L. Martinez-Martinez, H. Wang, A. Nohalez, E. Aizawa, P. Martinez-Redondo, A. Ocampo, P. Reddy, J. Roca, E. A. Maga, C. R. Esteban, W. T. Berggren, E. Nunez Delicado, J. Lajara, I. Guillen, P. Guillen, J. M. Campistol, E. A. Martinez, P. J. Ross, and J. C. Izpisua Belmonte. 2017. Interspecies chimerism with mammalian pluripotent stem cells. Cell 168(3):473–486.e415. https://doi.org/10.1016/j.cell.2016.12.036
- Yamaguchi, T., H. Sato, M. Kato-Itoh, T. Goto, H. Hara, M. Sanbo, N. Mizuno, T. Kobayashi, A. Yanagida, A. Umino, Y. Ota, S. Hamanaka, H. Masaki, S. T. Rashid, M. Hirabayashi, and H. Nakauchi. Interspecies organogenesis generates autologous functional islets. Nature 542(7640):191–196. https://doi.org/10.1038/nature21070
- Suzuki, K., C. Yu, J. Qu, M. Li, X. Yao, T. Yuan, A. Goebl, S. Tang, R. Ren, E. Aizawa, F. Zhang, X. Xu, R. D. Soligalla, F. Chen, J. Kim, N. Y. Kim, H. K. Liao, C. Benner, C. R. Esteban, Y. Jin, G. H. Liu, Y. Li, and J. C. Izpisua Belmonte. 2014. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell 15(1):31–36. https://doi.org/10.1016/j.stem.2014.06.016
- Porteus, M. 2015. Strategies to increase genome editing frequencies and to facilitate the identification of edited cells. Methods in Molecular Biology 1239:281–289. https://doi.org/10.1007/978-1-4939-1862-1_16
- Komor, A. C., A. H. Badran, and D. R. Liu. 2017. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell 168(1-2):20–36. https://doi.org/10.1016/j.cell.2016.10.044
- U.S. Food and Drug Administration. 2009. Guidance for industry: Q8(r2) pharmaceutical development. Available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q8r2-pharmaceutical-development (accessed August 25, 2020).
- Lipsitz, Y. Y., N. E. Timmins, and P. W. Zandstra. 2016. Quality cell therapy manufacturing by design. Nature Biotechnology 34(4):393–400. https://doi.org/10.1038/nbt.3525
- Alliance for Regenerative Medicine. 2014. Pharma and biotech survey. Available at: https://alliancerm.org/sites/default/files/ARM_Pharma_SurveyRept_Mar2014_e.pdf (accessed August 25, 2020).
- U.S. Environmental Protection Agency. 1995. National Technology Transfer and Advancement Act. Available at: https://www.epa.gov/laws-regulations/summary-national-technology-transfer-and-advancement-act#:~:text=The%20National%20Technology%20Transfer%20and%20Advancement%20Act%20(NTTAA)%20was%20signed,industrial%20innovation%20to%20the%20marketplace (accessed August 25, 2020).
